
An introduction to near infrared (NIR) spectroscopy
A.M.C. Davies
Norwich Near Infrared Consultancy, 10 Aspen Way, Cringleford, Norwich NR4 6UA, UK
When you hold your hand out to a burning fire you “feel” the heat being emitted by the fire but what is happening? The fire gives out light and infrared (IR) radiation; from a fire most of this is near infrared (NIR) radiation. Some of the NIR radiation is absorbed by water molecules in your skin. This raises the temperature of the water and results in an increase in temperature in the surrounding tissue which is detected by nerves in your skin. This radiation was discovered in 1800 by William Herschel, a musician and very successful amateur astronomer (he discovered the planet Uranus) because he wanted to know if any particular colour was associated with heat from sunlight. He found that the heat maximum was beyond the red end of the spectrum. Herschel could not believe that light and his “radiant heat” were related but he was wrong. By 1835 Ampere had demonstrated that the only difference between light and what he named “infrared radiation” was their wavelength. Then in 1864 James Maxwell wrote “This velocity [of electromagnetic force] is so nearly that of light that it seems we have strong reason to conclude that light itself (including radiant heat and other radiations) is an electromagnetic disturbance in the form of waves propagated through the electromagnetic field according to electromagnetic laws”. What we now call the electromagnetic spectrum is shown in Figure 1.

Early history of the study of infrared absorptions
The first (near) infrared spectra were measured in 1881 by Abney and Festing using photographic plates. Not only did they produce the first spectra but they also suggested, correctly, that the absorptions were related to the chemical composition of the liquids they investigated. The most important pioneer of IR spectroscopy was William W. Coblentz. In 1905 he published the result of a large study of compounds whose spectra he had recorded from 1000 nm to 16,000 nm. Coblentz’s work was a breakthrough in that researchers were able to relate the character of groups of atoms within molecules as being related to specific absorptions in the mid-IR (2500–50,000 nm). These absorptions are the result of interactions with the fundamental vibrations of the chemical bonds associated with the atoms of the groups. We can think of chemical bonds as weak springs holding together two or more atoms, these springs will vibrate naturally and when energy is added to the system then they will vibrate more energetically. However, atoms in molecules are constrained by quantum mechanics so that only a few specific energy levels are allowed. If we have only two atoms then the only vibration will be seen as a stretching. When three or more atoms are involved then bonds can also bend, giving rise to a whole series of different vibrations. Stretch vibrations require more energy than bending vibrations but there will also be variation in the energy requirements of the bending vibrations. Different chemical bonds (like O–H, C–H and N–H) vary in strength and hence the amount of energy required for the bond vibration to move from one level to the next. This variation in energy will be seen in a spectrum as a series of absorptions at different wavelengths. By looking at the spectrum we can deduce what vibrations are occurring and hence work out the structure of the molecule (or groups of atoms present).
One of the very useful properties of mid-IR spectra is that the region from 8500 nm to 12,500 nm is very characteristic for the molecule measured and this region is known as the “finger-print” region because it can be used to confirm the identity of many pure substances. While the study of mid-IR spectroscopy continued to grow, especially after World War II, interest in the NIR extended to quantitative measurements of water, a few simple organic compounds and a very few studies of specific proteins. No one considered it useful for characterising samples and it was considered too complex for use in quantitative analysis.
Absorptions in the NIR region
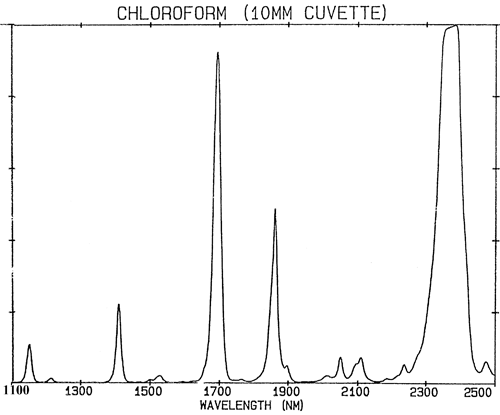
If chemical bonds behaved exactly like weak springs then quantum mechanics would restrict their vibration to just two states and there would be very few absorptions in the NIR region. Absorptions in the NIR region (780–2500 nm) are generated from fundamental vibrations by two processes; overtones and combinations. Overtones can be thought of as harmonics. So every fundamental will produce a series of absorptions at (approximately integer) multiples of the frequency (frequency is the reciprocal of wavelength). Combinations are rather more complex. NIR absorptions are at a higher state of excitement so they require more energy than a fundamental absorption. Combinations arise from the sharing of NIR energy between two or more fundamental absorptions. While the number of possible overtones from a group of fundamental absorptions in a molecule are limited to a few, a very large number of combinations will be observed. The effect of all these absorptions combine to make many NIR spectra to look rather uninteresting and to consist of only a few rather broad peaks. Figure 2 is an NIR spectrum of chloroform, CHCl3, the molecule contains only one hydrogen atom but all the absorption in its spectrum are caused by this single atom.
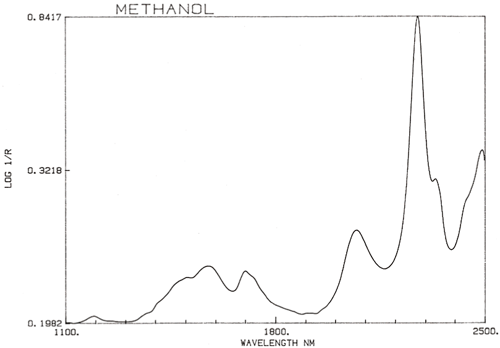
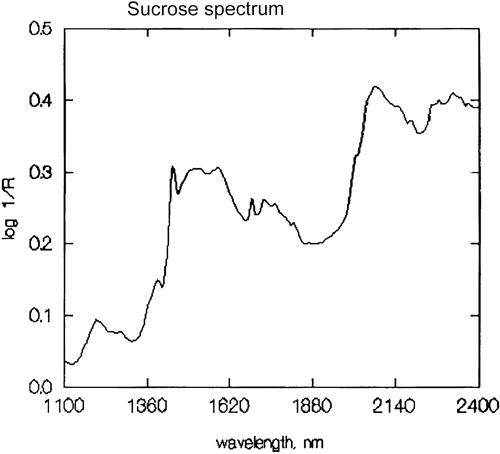
It is an important generalisation that NIR spectroscopy is dominated by hydrogen. Figure 3 is a spectrum of methanol, CH3OH, which contains four hydrogen atoms (but three are equivalent) and this spectrum is much more like a typical NIR spectrum with broad peaks. Figure 4 is a spectrum of sucrose, C12H24O12, which shows very broad areas of absorption but also some quite narrow peaks. It is important to realise that all of these broad absorptions are caused by multiple narrow, over-lapping absorptions. NIR spectra are much more complex than they appear.
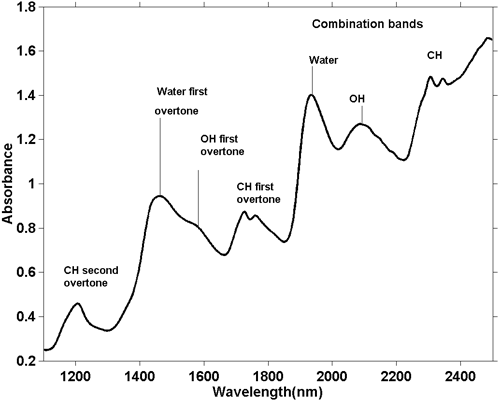
Although NIR spectra are more complicated it is possible to make some general observations. As a fundamental O–H stretching absorption is different to a fundamental C–H stretch then the series of overtones generated by these absorptions will also be different. The same goes for combination bands. The most common (and energetic) combination bands arise from stretch and bend combinations in the same group. So we see absorptions due the combination of O–H stretch with O–H bend and C–H stretch with C–H bend and these occur in different positions in the spectrum. Figure 5 is the NIR spectrum of a sample of biscuit dough. Biscuit dough contains several ingredients each of which contain many different molecules so this spectrum contains hundreds if not thousands of absorptions but we see the integration of them all and there appear to be just a few broad absorptions. From their position we can say in general terms the cause of the absorption, as indicated on the figure.
When the complexity of NIR absorption was first realised and compared to the relatively more easily understood mid-IR spectra, it was thought by most researchers that there was little to be gained by studying NIR spectroscopy. The region became neglected and students were wrongly instructed that there was nothing to be gained by studying the NIR region. Many students are still being taught the same opinion. The requirements were: very low noise spectrometers, the electronic computer, the application of mathematical techniques (chemometrics) and a genius to bring it all together. The man was Karl Norris; an engineer working for the USDA at Beltsville. He had not been taught spectroscopy so he did not know that there was nothing to be gained in the NIR region. So, rather like Herschel who looked for something where there was nothing, Norris developed the instruments and utilised computers to demonstrate that the NIR region was very useful for quantitative analysis, particularly of agricultural samples. One of the reasons why NIR analysis is so useful is that it can use reflected energy and this means that NIR analysis can be done with little or no sample preparation. Reflected energy is complex. First, because there are two components, specular (or mirror-like) and diffuse. In the context of NIR spectroscopy, the specular component does not give any information. The diffuse component depends on the physical nature of the sample; particle size being particularly important. Variation of the physical parameters of a sample causes changes in the spectrum so that the observed spectrum is a mixture of chemical and physical information.
The use of reflected energy was forced on Karl Norris. While it makes possible the NIR analysis of a very much wider range of samples, it does add another layer of complication. A complete mathematical theory of reflection spectroscopy is not yet available but it has been found possible by good experimental practice and the utilisation of mathematical techniques to use NIR reflection spectroscopy for analytical chemistry. Because the technique can be applied with little or no sample preparation, analysis times are reduced from hours to minutes and furthermore several analytical results can be obtained from the same NIR data while the conventional analysis would often require another technique and more hours of work. It is, however, necessary to develop calibrations which require many samples, many hours of work and thousands (or probably millions) of computer calculations. With these sorts of attributes it is not surprising that 40 years after the ground-breaking research, a very wide range of analysis can be achieved by NIR spectroscopy.
What is surprising is that in spite of the success of NIR spectroscopic analysis, world-wide there are very few university chemistry departments that have any programme of research in NIR spectroscopy. Consequently, the majority of chemistry students leave university with no knowledge of NIR, with the possible exception of the old fashioned view that there is nothing useful to learn about the NIR region.